Leave message
Can’t find what you’re looking for?
Fill out this form to inquire about our custom protein services!
Inquire about our Custom Services >>






























> Insights > Sonderthema zur tiefgreifenden Interpretation der Qualität von GMP-Produkten - Thema 3

In den letzten Jahren hat die biopharmazeutische Industrie ein bemerkenswertes Wachstum erlebt, wobei sich die Zell- und Gentherapie inmitten des kontinuierlichen technologischen Fortschritts als ein zentraler Sektor herauskristallisiert hat. Da Biologika einen immer größeren Anteil an der Medizin der nächsten Generation einnehmen, hat die Gewährleistung der Sicherheit dieser biologischen Arzneimittel oberste Priorität. Dies führt dazu, dass nicht nur die endgültige Zell- und Gentherapie, sondern auch die bei der Herstellung und Produktion verwendeten Rohstoffe einer genaueren Prüfung unterzogen werden. Dies wurde noch dadurch unterstrichen, dass die FDA die Verwendung geeigneter Materialien „von höchster Qualität“ betonte. In ähnlicher Weise hat die International Pharmaceutical Regulatory Plan Cell Therapy Working Group ein beratendes Dokument verfasst, in dem sie ihren Standpunkt zur Verwendung hochwertiger Rohstoffe bei der Herstellung und Zulassung humaner CGT-Produkte darlegt.
Was bedeutet das für die Hersteller von Zelltherapien, wenn die Aufsichtsbehörden die Verwendung von „hochwertigen“ Rohstoffen betonen?
Kurzum, die Hersteller von Zelltherapien müssen die Qualität ihrer Rohstoffe sorgfältig prüfen, unabhängig davon, ob sie von einem Zulieferer stammen oder selbst entwickelt wurden.
Bewertungen der Qualitätskontrollmethoden und -standards für relevante Rohstoffe, einschließlich Zytokine und andere Wachstumsfaktoren, sind ein notwendiger Schritt im Rahmen eines IND-Antrags (Investigational New Drug), bevor mit der klinischen Forschung begonnen wird. Diese Methoden und Standards stammen aus verschiedenen Quellen. Pharmakopöe-Methoden, die von Organisationen wie der USP oder dem European Directorate for the Quality of Medicines & Healthcare (EDQM) abgeleitet sind, sind standardisierte Ansätze, die eine grundlegende Anforderung für die Produktion und Freigabe festlegen. Nicht-pharmakopische Methoden, wie die Dokumente der Internationalen Harmonisierungskonferenz (ICH), sind ergänzende analytische Ansätze, die auf die Bewertung spezifischer Merkmale eines Produkts und seines Herstellungsprozesses zugeschnitten sind.
Viele kommerzielle Produkte werden derzeit nach diesen Normen vermarktet, die gemeinhin als „GMP- oder cGMP-grade“ bezeichnet werden. Produkte, die den genannten Normen entsprechen, sind jedoch häufig selbst reguliert. Die Hersteller von Zelltherapien sind daher nach wie vor für die Durchführung einer Due-Diligence-Prüfung verantwortlich, was es schwierig machen kann, einen vertrauenswürdigen Lieferanten zu finden.
Im Folgenden werden die drei Hauptaspekte eines GMP-gerechten Qualitätskontrollsystems für Rohstoffe hervorgehoben, die für die Erfüllung der IND-Anforderungen und den erfolgreichen Eintritt in die klinische Phase entscheidend sind.
Die Bewertung der Sicherheit von Rohstoffen wird durch verschiedene Kontaminationskontrollen und Nachweisstrategien definiert, um Rückstände zu begrenzen, die Schäden verursachen oder das endgültige Therapeutikum beeinträchtigen könnten. Dazu gehören Sterilitätstests, Bewertungen von Verunreinigungen wie Endotoxinen, Mykoplasmen, Rückstände der Wirtszell-DNA und Rückstände von Wirtszellproteinen.
• USP <71> Sterilitätstests
Die Gewährleistung der Sterilität der endgültigen Zell- und Gentherapien ist schwierig. Da es sich bei dem endgültigen Therapeutikum um Zellen handelt, gibt es keine einfache Methode wie sterile Filtration, Autoklavieren usw. Daher müssen die Hersteller während des gesamten Herstellungsprozesses sehr vorsichtig sein, um die Sterilität des Endprodukts zu gewährleisten. Dies gilt auch für die verwendeten Rohstoffe, so dass die Hersteller ihre Zulieferer genauestens prüfen müssen. Daher wird erwartet, dass die Rohstofflieferanten für GMP-konforme Hersteller, zu denen auch wir gehören, Zytokine und andere Wachstumsfaktoren streng kontrollieren, indem sie eine automatische Abfüllung in B+A-Reinräumen, Online-Umgebungsmonitore und USP-konforme Sterilitätstests einsetzen. Somit ist die Bewertung der Sterilität von Rohstoffen die wichtigste Methode zur Gewährleistung der Sterilität des endgültigen Therapeutikums. Weitere Informationen finden Sie in unserem Spezialthema über die tiefgreifende Interpretation der GMP-Produktqualität - Thema 2.
• USP <85> Endotoxin-Tests
Endotoxine sind in Bakterienzellen enthaltene Toxine, die nach dem Zerfall der Zellen freigesetzt werden und manchmal zu Krankheiten wie Botulismus führen können. Die Aufrechterhaltung eines niedrigen Endotoxingehalts in Biologika ist für die Gewährleistung der Patientensicherheit und die Vorbeugung endotoxinbedingter Krankheiten von wesentlicher Bedeutung. Es sind mehrere Methoden zulässig: qualitative Gelgerinnung und chromogene LAL-Methoden. Gibt es jedoch uneinheitliche oder widersprüchliche Ergebnisse, so gilt das Ergebnis der Gelbildung als maßgebend. Intern verwenden wir eine LAL-Methode, die anhand von USP-Referenzstandard-Endotoxin-Kalibratoren kalibriert wird, um die Genauigkeit der in Tabelle 1 aufgeführten Probenergebnisse zu gewährleisten. Um sicherzustellen, dass die Endotoxin-Testmethode gültig ist, wurden anschließend vorbereitende Tests gemäß USP <85> durchgeführt, wobei eine akzeptable Endotoxin-Wiederfindungsrate im Bereich von 50 bis 200 % definiert wurde.
Table 1. Endotoxin Sample Curve Evaluation
| Theoretical value | Detected value | Recovery rate |
|---|---|---|
| 50 EU/ml | 55.04 EU/ml | 113.0% |
| 5 EU/ml | 5.96 EU/ml | 104.7% |
| 0.5 EU/ml | 0.6 EU/ml | 108.5% |
| 0.05 EU/ml | 0.06 EU/ml | 108.0% |
• USP <63> Mykoplasmen-Tests
Mykoplasmen sind eine häufige Verunreinigung in Zell- und Gewebekulturen, die zu einer Veränderung des Zellwachstums und des Phänotyps führt. Die Prüfung auf Mykoplasmen ist von entscheidender Bedeutung für die Gewährleistung der Zuverlässigkeit von Biologika und ihren Rohstoffen. Der Goldstandard bei der Bewertung von Mykoplasmen ist nach wie vor die Kultivierungsmethode, bei der das Wachstum typischer Mykoplasmenkolonien auf festen Medien überwacht wird. Andere validierte Methoden umfassen die Verwendung von Fluoreszenzfarbstoffen zur Hervorhebung charakteristischer Partikel oder fadenförmiger Muster auf der Zelloberfläche. Obwohl auch Nukleinsäure-Amplifikationstechniken oder auf Enzymaktivität basierende Methoden verwendet werden können, müssen auch eine geeignete Validierung und Vergleiche zwischen den Methoden mit Zellkulturen angesprochen werden.
• USP <509, 1132> Rückstände von Wirtszell-DNA und Proteinen
Wirtszell-DNA und -Protein sind weitere Faktoren, die ebenfalls auf akzeptable Werte kontrolliert werden müssen, um Sicherheitsrisiken zu vermeiden. Fremde DNA und Proteine können immunogene und onkogene Reaktionen auslösen, die noch mehr Schaden anrichten können. Für die Sicherheit der Patienten ist es von entscheidender Bedeutung, dass diese Verunreinigungen in den Rohstoffen und den nachfolgenden Herstellungsprozessen entfernt werden.
Natürlich ist wie bei jeder Test- oder Analysemethode eine Validierung erforderlich, um sicherzustellen, dass die verwendeten Tests korrekt sind. Die oben erwähnten USP-Dokumente enthalten Testprotokolle und klare Kriterien für die Bewertung von Tests, die für alle biologischen Materialien standardisiert sind. Wenn es jedoch um stoffspezifische Bewertungen wie die Bioaktivität geht, müssen die Analysemethoden zunächst validiert werden, was in den meisten Fällen nach ICH Q2 (Revision 1) mit dem Titel „Validation of Analytical Procedures Text and Methodology“ erfolgt.
Table 2. Validation Characteristics for Consideration
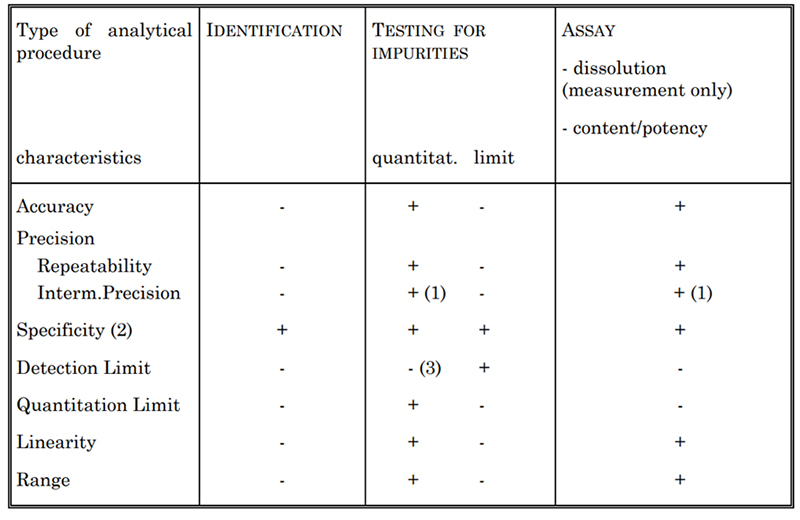
Je nach Art des zu validierenden Assays werden im Allgemeinen bestimmte Merkmale bewertet, die in Tabelle 2 aufgeführt sind. Dazu gehören Spezifität, Genauigkeit, Präzision, Nachweisgrenze, Quantifizierungsgrenze, Linearität und Bereich. Zur Bestimmung der Zytokinkonzentration setzen wir beispielsweise verschiedene Testmethoden ein, wie den US-spektrophotometrischen Test und die Lowry-Methode. Jede dieser Methoden ist gemäß den USP- und ICH Q2-Richtlinien vollständig validiert.
Table 3. Validation of UV-Spectrophotometric Assay
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Accuracy | Recovery Rate: 96% | Recovery Rate: 90%-108% | Pass |
| Repeatability | RSD: 0.05% | RSD≤3% | Passtd> |
| Intermediate Precision | RSD: 0.48% | RSD≤3% | Pass |
| Robustness | RSD: 0.04% | RSD≤3% | Pass |
| Linearity | R2: 0.99925 | R2>0.999 | Pass |
| Range | 0.0452-0.452 mg/ml | 0.0452-0.452 mg/ml | Pass |
Zur weiteren Validierung des UV-spektrophotometrischen Assays wurde eine zweite Bestätigungsmethode, die Lowry-Methode, verwendet. Als ausgereifte, gut etablierte Methode bietet die Lowry-Methode eine solide Grundlage für den Vergleich. Direkte Vergleiche der quantitativen Ergebnisse von sechs verschiedenen aufgestockten Proben gewährleisten somit die Genauigkeit und Zuverlässigkeit einer UV-Methode (siehe Tabelle 4).
Table 4. Secondary Confirmation Assay – Lowry Method
| Method Validation | Lowry | UV Method | Conclusion |
|---|---|---|---|
| Quantitative Result | 461 ug/ml | 436 ug/ml | Pass |
| 476 ug/ml | 436 ug/ml | ||
| 465 ug/ml | 436 ug/ml | ||
| 472 ug/ml | 436 ug/ml | ||
| 468 ug/ml | 436 ug/ml | ||
| 461 ug/ml | 435 ug/ml |
Unter Verwendung desselben Rahmens für die Validierung von Analysemethoden umfasst dies auch andere Tests wie die Zellaktivität. Unser TNF-α-Assay umfasst zum Beispiel Genauigkeit, mittlere Präzision, Linearität und Bereich. Die Kriterien unterscheiden sich jedoch je nach dem Zweck des Tests.
Table 5 Example of TNF-α Cell activity validation
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Relative Accuracy |
Bias:1.1% Slope:1.01 |
Bias within ±12% range & Slope of regression equation between 0.80 and 1.25 | Pass |
| Intermediate Precision | GCV*:11.2% | (GCV)* ≤ 20% | |
| Specificity | Difference % (buffer): 8.8% | Difference % (buffer) ≤ ±10% | |
| Linearity | Correlation Coefficient: 0.97 | Correlation Coefficient ≥ 0.95 | |
| Potency Range | Potency Range: 64% -156% | Range of product efficacy standards (64% -156%) |
*GCV is calculated as the anti-log of the standard deviation. GCV = anti-log(SD)
Stabilitätsprüfungen sind die Grundlage für die Bewertung der Haltbarkeit eines Produkts. Daher sind diese Tests ein entscheidender Bestandteil der gesamten Arzneimittelentwicklung, der klinischen Entwicklung, der Markteinführung und der Überwachung nach der Markteinführung im Rahmen von Qualitätsforschungsprozessen. Das bedeutet, dass die Stabilitätsprüfung auf der Grundlage der einzigartigen Eigenschaften und Merkmale des Produkts durchgeführt werden muss. Darüber hinaus gewährleistet die systematische Durchführung von Stabilitätsstudien minimale Schwankungen von Charge zu Charge und liefert gleichzeitig klare Anweisungen für die Lagerung bei der Lieferung.
Um die Stabilität und Konsistenz innerhalb eines überschaubaren Zeitrahmens zu bewerten, ist die Methode der beschleunigten Stabilitätsprüfung auf der Grundlage der Arrhenius-Gleichung eine weithin anerkannte Strategie, deren Anwendbarkeit und Genauigkeit in den letzten Jahren in zahlreichen Studien nachgewiesen wurde. Sowohl das 2002 vom Europäischen Komitee für Normung veröffentlichte Dokument „EN13640 In Vitro Diagnostic Medical Devices Stability Testing“ als auch das 2009 vom Clinical and Laboratory Standards Institute (CLSI) herausgegebene Dokument „EP25A Evaluation of Stability of In Vitro Diagnostic Reagents“ empfehlen die Anwendung dieser Methode zur Bestimmung der Stabilität von In-Vitro-Diagnostik-Reagenzien. Die verschiedenen Parameter der Stabilitätsbewertung sind in Tabelle 6 aufgeführt.
Table 6. Stability Assessment Parameters

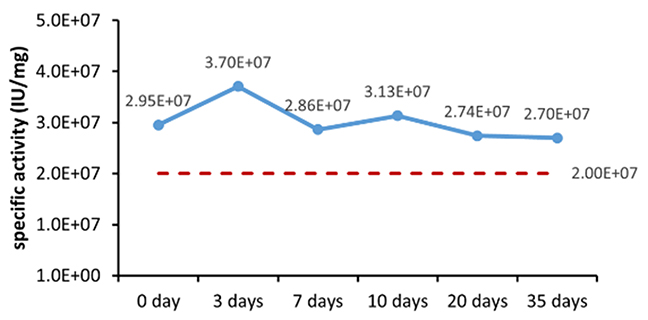
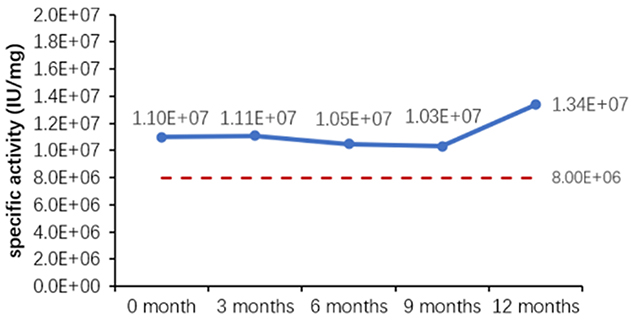
Figure 1. (A) Accelerated and (B) real-time stability evaluations of finished product (powder).
QDie Qualitätskontrolle entscheidet sich im Allgemeinen für reifere und empfindlichere Produkte, die den festgelegten Anforderungen entsprechen. Aufgrund der potenziellen Einschränkungen bei den Analysemethoden ist es ratsam, den Einsatz gleichzeitiger ergänzender Methoden in Betracht zu ziehen, um nicht nur die Rohstoffe, sondern auch das fertige Therapeutikum umfassend zu bewerten. Auch die Analysemethoden selbst müssen einer strengen Validierung unterzogen werden. Ein gut eingeführtes, strenges und professionelles Qualitätskontrollsystem kann über Erfolg oder Misserfolg eines therapeutischen Zulassungsverfahrens entscheiden und sollte sich auf Pharmakopöe-Normen wie die USP beziehen. So hat sich die internationale Sichtweise auf das pharmazeutische Qualitätsmanagement von „Arzneimittelqualität wird durch Inspektion kontrolliert“ zu „Arzneimittelqualität wird durch Prozesskontrolle in der Produktion erreicht“ und schließlich zu „Arzneimittelqualität wird durch gutes Design (Quality by Design, QbD) erzeugt“ entwickelt Die Umsetzung dieser QbD-Philosophie setzt voraus, dass eine Korrelation zwischen den Qualitätsmerkmalen des Produkts und der klinischen Sicherheit und Wirksamkeit hergestellt wird, was die Schaffung eines umfassenden Qualitätskontrollsystems während des gesamten Prozesses und des Produktlebenszyklusmanagements erfordert. Aus diesem Grund entwickelt ACROBiosystems sein GMP-Qualitätsmanagementsystem kontinuierlich weiter, um eine zuverlässige Unterstützung bei der Beschaffung von Rohstoffen für Zell- und Gentherapien zu bieten.



This web search service is supported by Google Inc.
















 A-Z
A-Z