Leave message
Can’t find what you’re looking for?
Fill out this form to inquire about our custom protein services!
Inquire about our Custom Services >>






























> Insights > Thème spécial sur l'interprétation approfondie de la qualité des produits selon les bonnes pratiques de fabrication (BPF/GMP) - Thème 3

Ces dernières années, l'industrie biopharmaceutique a connu une croissance remarquable, la thérapie cellulaire et génique s'imposant comme un secteur clé dans un contexte de progrès technologiques constants. À mesure que les produits biologiques prennent une place de plus en plus importante dans la médecine de nouvelle génération, la sécurité de ces médicaments biologiques est devenue une priorité absolue. Cela a pour conséquence une attention accrue tournée non seulement sur la thérapie cellulaire et génique finale, mais aussi sur les matières premières utilisées lors de la fabrication et de la production. La FDA a d'ailleurs souligné l'importance de l'utilisation de matériaux appropriés de "la plus haute qualité". De même, le groupe de travail sur la thérapie cellulaire du Plan international de réglementation pharmaceutique a rédigé un document délibératif diffusant son point de vue sur l'utilisation de matières premières de haute qualité dans la fabrication et l'homologation de produits de CGT humaine.
Les organismes de réglementation mettent l'accent sur l'utilisation de matières premières de "haute qualité", qu'est-ce que cela signifie pour les fabricants de thérapies cellulaires ?
En bref, les fabricants de thérapies cellulaires doivent faire preuve de diligence raisonnable pour garantir la qualité de leurs matières premières, qu'elles proviennent d'un fournisseur ou qu'elles soient développées en interne.
L'évaluation des méthodes et des normes de contrôle de la qualité des matières premières pertinentes, y compris les cytokines et autres facteurs de croissance, est une étape nécessaire dans le cadre d'une demande de nouveau médicament de recherche (IND) avant de passer à la recherche clinique. Ces méthodes et normes proviennent de plusieurs sources. Les méthodesde la pharmacopée, dérivées d'organisations telles que l'USP ou la Direction européenne de la qualité du médicament et des soins de santé (EDQM), sont des approches normalisées qui établissent une exigence fondamentale pour la production et la libération. Les méthodes hors pharmacopée, telles que les documents de la Conférence internationale sur l'harmonisation (ICH), sont des approches analytiques complémentaires conçues pour évaluer des caractéristiques spécifiques d'un produit et de son processus de fabrication.
De nombreux produits commerciaux sont actuellement commercialisés dans le respect de ces normes, communément appelés "qualité GMP" ou "qualité cGMP". Toutefois, les produits qui respectent les normes énoncées sont souvent autoréglementés. En tant que tel, il incombe toujours aux fabricants de thérapies cellulaires de faire preuve de diligence, ce qui peut rendre difficile la recherche d'un fournisseur digne de confiance.
Nous soulignons ici les trois principaux aspects d'un système de contrôle de la qualité des matières premières conforme aux GMP, qui sont essentiels pour répondre aux exigences des IND et passer avec succès à la phase clinique.
L'évaluation de la sécurité des matières premières est définie par divers contrôles des contaminants et diverses stratégies de détection afin de limiter tout résidu susceptible de causer des dommages ou d'affecter le produit thérapeutique final. Cela comprend des tests de stérilité, des évaluations de contaminants tels que les endotoxines, les mycoplasmes, l'ADN résiduel des cellules hôtes et les protéines résiduelles des cellules hôtes.
• USP <71> Tests de stérilité
Il est difficile de garantir la stérilité des thérapies cellulaires et géniques finales. Comme le produit thérapeutique final est constitué de cellules, il n'existe pas de méthode simple telle que la filtration stérile, le passage à l'autoclave, etc. Les fabricants doivent donc être très vigilants tout au long du processus de fabrication pour garantir la stérilité du produit final. Cela vaut également pour les matières premières utilisées, ce qui oblige les fabricants à évaluer rigoureusement leurs fournisseurs. Ainsi, on attend des fournisseurs de matières premières pour les fabricants conformes aux GMP, dont nous faisons partie, un contrôle rigoureux des cytokines et autres facteurs de croissance, en incorporant des systèmes de remplissage et de finition automatiques dans des salles blanches B+A, des moniteurs environnementaux en ligne et des tests de stérilité conformes à l'USP. Ainsi, l'évaluation de la stérilité des matières premières est la principale méthode pour garantir la stérilité du produit thérapeutique final. Pour plus d'informations, lisez notre thème spécial sur l' interprétation approfondie des GMP en matière de qualité des produits - thème 2.
• USP <85> Tests d'endotoxine
Les endotoxines sont des toxines présentes dans les cellules bactériennes, elles sont libérées après la désintégration des cellules, ce qui peut parfois entraîner des maladies telles que le botulisme. Le maintien d'un faible taux d'endotoxines dans les produits biologiques est essentiel pour garantir la sécurité des patients et prévenir les maladies liées aux endotoxines. Plusieurs méthodes sont acceptables : les méthodes qualitatives à base de gel et les méthodes chromogènes à base de LAL. Toutefois, en cas de résultats incohérents ou contradictoires, c'est le résultat du caillot de gel qui fait foi. En interne, nous utilisons une méthode LAL étalonnée par rapport aux étalons d'endotoxines de référence USP pour garantir la précision; des exemples de résulats sont présentés dans le tableau 1. Ensuite, pour s'assurer de la validité de la méthode de test des endotoxines, des tests préparatoires ont été effectués conformément à la norme USP <85>, un taux de récupération des endotoxines acceptable étant défini comme se situant dans une fourchette de 50 à 200 %.
Table 1. Endotoxin Sample Curve Evaluation
| Theoretical value | Detected value | Recovery rate |
|---|---|---|
| 50 EU/ml | 55.04 EU/ml | 113.0% |
| 5 EU/ml | 5.96 EU/ml | 104.7% |
| 0.5 EU/ml | 0.6 EU/ml | 108.5% |
| 0.05 EU/ml | 0.06 EU/ml | 108.0% |
• USP <63> Tests sur les mycoplasmes
Les mycoplasmes sont des contaminants courants dans les cultures de cellules et de tissus qui entraînent une altération de la croissance et du phénotype cellulaires. La recherche de mycoplasmes est essentielle pour garantir la fiabilité des produits biologiques et de leurs matières premières. Lors de l'évaluation des mycoplasmes, l'étalon-or reste la méthode de culture, qui consiste à surveiller la croissance de colonies typiques de mycoplasmes sur des milieux solides. D'autres méthodes validées incluent l'utilisation de colorants fluorescents pour mettre en évidence des particules caractéristiques ou des motifs filamenteux à la surface des cellules. Bien que des techniques d'amplification de l'acide nucléique ou des méthodes basées sur l'activité enzymatique puissent également être utilisées, il convient également d'aborder la question de la validation et des comparaisons entre les méthodologies et la culture cellulaire.
• USP <509, 1132> ADN et protéines résiduelles de la cellule hôte
L'ADN et les protéines de la cellule hôte sont d'autres facteurs qui doivent également être contrôlés à des niveaux acceptables afin d'éviter tout risque pour la sécurité. L'ADN et les protéines étrangers sont susceptibles de provoquer des réactions immunogènes et oncogènes qui peuvent être plus dommageables. Il est essentiel pour la sécurité des patients de veiller à ce que ces impuretés soient éliminées des matières premières et des processus de fabrication ultérieurs.
Bien entendu, comme pour toute méthode de test ou d'analyse, une validation est nécessaire pour s'assurer que les tests utilisés sont corrects. Les documents USP mentionnés ci-dessus fournissent des protocoles de test et des critères clairs pour l'évaluation des tests qui sont standardisés pour tous les matériaux biologiques. Cependant, lorsqu'il s'agit d'évaluations spécifiques à un matériau, telles que la bioactivité, les méthodes analytiques doivent d'abord être validées, le plus souvent conformément à la norme ICH Q2 (révision 1), intitulée "Validation of Analytical Procedures Text and Methodology" (validation du texte et de la méthodologie des procédures analytiques).
Table 2. Validation Characteristics for Consideration
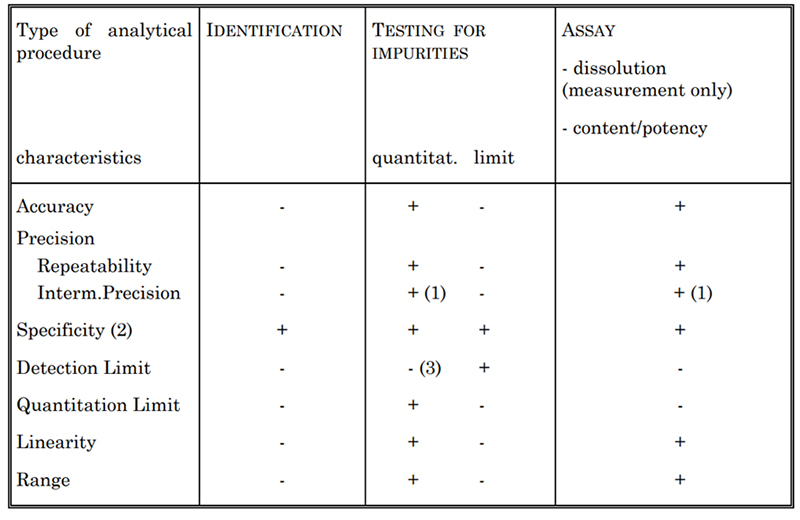
Selon le type de test à valider, certaines caractéristiques sont généralement évaluées, comme indiqué dans le tableau 2. Elles comprennent la spécificité, l'exactitude, la précision, la limite de détection, la limite de quantification, la linéarité et l'étendue. Par exemple, pour déterminer la concentration de cytokines, nous établissons différentes méthodes de dosage, telles que le dosage spectrophotométrique US et la méthode Lowry. Chacune de ces méthodes est entièrement validée conformément aux directives USP et ICH Q2.
Table 3. Validation of UV-Spectrophotometric Assay
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Accuracy | Recovery Rate: 96% | Recovery Rate: 90%-108% | Pass |
| Repeatability | RSD: 0.05% | RSD≤3% | Passtd> |
| Intermediate Precision | RSD: 0.48% | RSD≤3% | Pass |
| Robustness | RSD: 0.04% | RSD≤3% | Pass |
| Linearity | R2: 0.99925 | R2>0.999 | Pass |
| Range | 0.0452-0.452 mg/ml | 0.0452-0.452 mg/ml | Pass |
Pour valider davantage le test spectrophotométrique UV, une méthode de confirmation secondaire, la méthode Lowry, a été utilisée. En tant que méthodologie mature et bien établie, la méthode Lowry constitue une base de comparaison solide. Ainsi, les comparaisons directes des résultats quantitatifs de six échantillons enrichis différents garantissent la précision et la fiabilité d'une méthode UV, comme le montre le tableau 4.
Table 4. Secondary Confirmation Assay – Lowry Method
| Method Validation | Lowry | UV Method | Conclusion |
|---|---|---|---|
| Quantitative Result | 461 ug/ml | 436 ug/ml | Pass |
| 476 ug/ml | 436 ug/ml | ||
| 465 ug/ml | 436 ug/ml | ||
| 472 ug/ml | 436 ug/ml | ||
| 468 ug/ml | 436 ug/ml | ||
| 461 ug/ml | 435 ug/ml |
En utilisant le même cadre de validation de la méthode analytique, ceci inclut d'autres tests tels que l'activité cellulaire. Par exemple, notre test TNF-α comprend l'exactitude, la précision intermédiaire, la linéarité et l'étendue. Toutefois, les critères diffèrent en fonction de l'objectif du test.
Table 5 Example of TNF-α Cell activity validation
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Relative Accuracy |
Bias:1.1% Slope:1.01 |
Bias within ±12% range & Slope of regression equation between 0.80 and 1.25 | Pass |
| Intermediate Precision | GCV*:11.2% | (GCV)* ≤ 20% | |
| Specificity | Difference % (buffer): 8.8% | Difference % (buffer) ≤ ±10% | |
| Linearity | Correlation Coefficient: 0.97 | Correlation Coefficient ≥ 0.95 | |
| Potency Range | Potency Range: 64% -156% | Range of product efficacy standards (64% -156%) |
*GCV is calculated as the anti-log of the standard deviation. GCV = anti-log(SD)
Les tests de stabilité constituent la base de l'évaluation de la durée de conservation d'un produit. En tant que tel, ce test est un élément essentiel tout au long du développement du médicament, de la phase clinique, de la mise sur le marché et de la surveillance après la mise sur le marché dans le cadre des processus de recherche sur la qualité. Cela signifie que les tests de stabilité doivent être effectués en fonction des qualités et des caractéristiques uniques du produit. En outre, la réalisation systématique d'études de stabilité permet de réduire au minimum les variations d'un lot à l'autre, tout en fournissant des instructions claires pour le stockage lors de la livraison.
Pour évaluer la stabilité et la cohérence dans un délai viable, la méthode de test de stabilité accélérée, basée sur l'équation d'Arrhenius, est une stratégie largement reconnue, et de nombreuses études réalisées ces dernières années ont démontré son applicabilité et sa précision. Le document "EN13640 In Vitro Diagnostic Medical Devices Stability Testing" publié en 2002 par le Comité européen de normalisation et le document "EP25A Evaluation of Stability of In Vitro Diagnostic Reagents" publié en 2009 par le Clinical and Laboratory Standards Institute (CLSI) recommandent tous deux l'utilisation de cette méthode pour déterminer la stabilité des réactifs de diagnostic in vitro. Les différents paramètres d'évaluation de la stabilité sont présentés dans le tableau 6.
Table 6. Stability Assessment Parameters

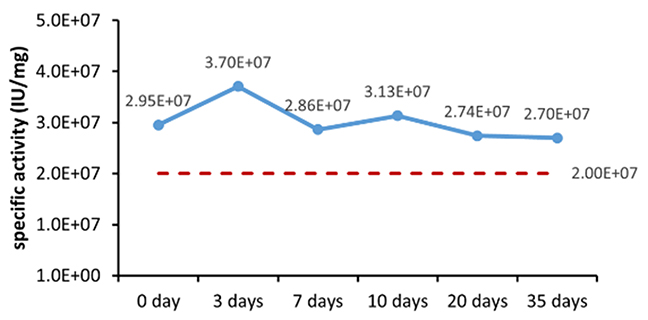
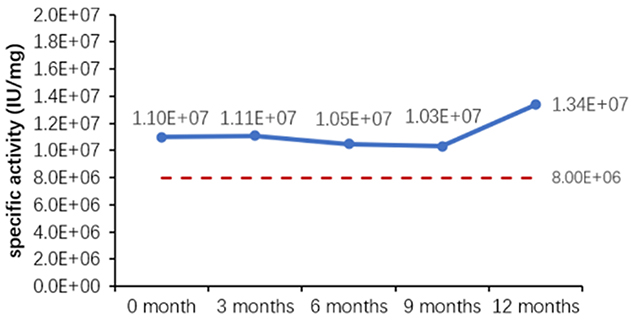
Figure 1. (A) Accelerated and (B) real-time stability evaluations of finished product (powder).
Les évaluations de contrôle de la qualité optent généralement pour des produits plus mûrs et plus sensibles qui répondent aux exigences spécifiées. En raison des limites potentielles des méthodes analytiques, il est conseillé d'envisager l'utilisation simultanée de méthodes complémentaires afin d'évaluer de manière exhaustive non seulement les matières premières, mais aussi le produit thérapeutique final. Les méthodes analytiques elles-mêmes doivent également faire l'objet d'une validation rigoureuse. Un système de contrôle de la qualité bien établi, rigoureux et professionnel peut être l'élément déterminant de tout processus d'approbation thérapeutique et doit faire référence aux normes de la pharmacopée telles que l'USP. Ainsi, la perspective internationale de la gestion de la qualité pharmaceutique a évolué, passant de "la qualité des médicaments est contrôlée par l'inspection" à "la qualité des médicaments est obtenue par le contrôle des processus de production" et, enfin, à "la qualité des médicaments est obtenue par une bonne conception (qualité par conception, Quality by design, QbD)". L'adoption de cette philosophie QbD implique l'établissement d'une corrélation entre les attributs de qualité du produit et la sécurité et l'efficacité cliniques, ce qui nécessite la création d'un système complet de contrôle de la qualité tout au long du processus et de la gestion du cycle de vie du produit. C'est dans cet état d'esprit qu'ACROBiosystems perfectionne en permanence son système de gestion de la qualité GMP afin de fournir un soutien fiable en matière de matières premières pour les thérapies cellulaires et géniques.



This web search service is supported by Google Inc.
















 A-Z
A-Z