Leave message
Can’t find what you’re looking for?
Fill out this form to inquire about our custom protein services!
Inquire about our Custom Services >>






























> Insights > Special Topic on Deep Interpretation of GMP Product Quality--Topic 1

The quality of materials and reagents utilized in the production process of Cell and Gene Therapy (CGT) plays a pivotal role in determining the ultimate product quality and therapeutic safety. This is particularly crucial in safeguarding against exogenous contamination, which encompasses inadvertent introduction of contaminants like bacteria, fungi, mycoplasma, and viruses into raw materials, cell substrates, and production products Generally, sponsors should ensure appropriate quality control of materials and reagents to mitigate unreasonable risks to subjects or patients, such as implementing sterilization or sterility testing to ensure the absence of microbial and viral contaminants. Stringent quality control of raw materials used in CGT production is a necessary measure to reduce the risk of exogenous factor contamination in CGT final products and ensure the safety and efficacy of CGT products.
To ensure the safety of upcoming therapeutics, regulatory agencies across the globe have established relevant requirements regarding the safety of raw materials used in CGT production. In particular, the FDA recommends the use of readily available and feasible materials and reagents of the highest quality for CGT production. These materials and reagents are often labeled as "GMP-grade" or claimed for use in cell therapy production. Key regulatory documents include:
(1) The "Content and Review of Chemistry, Manufacturing, and Controls (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications" issued by FDA in April 2008.
(2) USP<1043>"Ancillary Materials for Cell, Gene, and Tissue-Engineered Products".
(3) EP 5.2.12 "Raw Materials of Biological Origin for the Production of Cell and Gene Therapy Medicinal Products".
All these three regulatory documents mentioned the safety of raw materials and emphasized the importance of safety.
Our GMP-grade products have been designed and developed with a comprehensive consideration of safety, including the control of exogenous factor contamination throughout the entire production and quality control process. Here are the key aspects:
1. The cell line used for production is well-documented and traceable to ECACC as the source.
2. The production host cells, HEK293, after domestication and establishment, undergo comprehensive testing (26 items).
The testing is conducted by an internationally renowned third-party testing organization, in accordance with the following regulations:
• ICH Q5 Part III: Cell Line Characterization: viral testing.
• FDA's "Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals" (1993), specifically the "V. QUALITY CONTROL TESTING" section.
• FDA's "Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications" (2010).
3. The engineered cells undergo further testing after the construction of the cell bank. The testing, conducted by a well-known domestic third-party testing organization, includes sterility, mycoplasma, cell morphology, different indicator cell inoculation methods in vitro, retrovirus and exogenous virus detection, among others.
4. The upstream cell culture stage uses a chemically defined medium (CDM) as a well-defined culture medium. All materials used in the upstream production process are serum-free (SF) and animal-origin-free (AOF). Cell recovery and amplification are performed in a Class C clean area biosafety cabinet (BSC). The reactor production stage utilizes disposable closed systems and aseptic welding technology. Different stages of production are separated to minimize the risk of contamination and cross-contamination.
5. During downstream purification process design, virus removal/inactivation steps such as low pH treatment and nanofiltration are introduced. Viral removal process validation is also conducted (in progress) for critical steps. The purification process utilizes project-specific separation columns and chromatographic media to avoid cross-contamination risks between different production stages.
The following regulations provide guidance on viral safety:
• Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. 1999. International Conference on Harmonization(ICH)Q5A(R1).
• Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products. EMEA /CHMP / BWP / 398498 / 2005. European Medicines Agency (EMA). 2009.
• Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal. USP General Chapters:<1050>.
6. The formulation production is carried out in a strict Grade B+A production environment. After sterilizing filtration, the semi-finished product is processed using fully automated equipment and a single-use sterile filling system. Operations such as aseptic component assembly, filling, stoppering, automatic infeed and outfeed, and lyophilization are performed in a B+A-grade clean environment (monitored using PMS continuous environmental monitoring system). After lyophilization, vials are sealed in a Grade C+A environment. The finished products undergo manual inspection, labeling, and other operations before being released after passing the quality inspection.
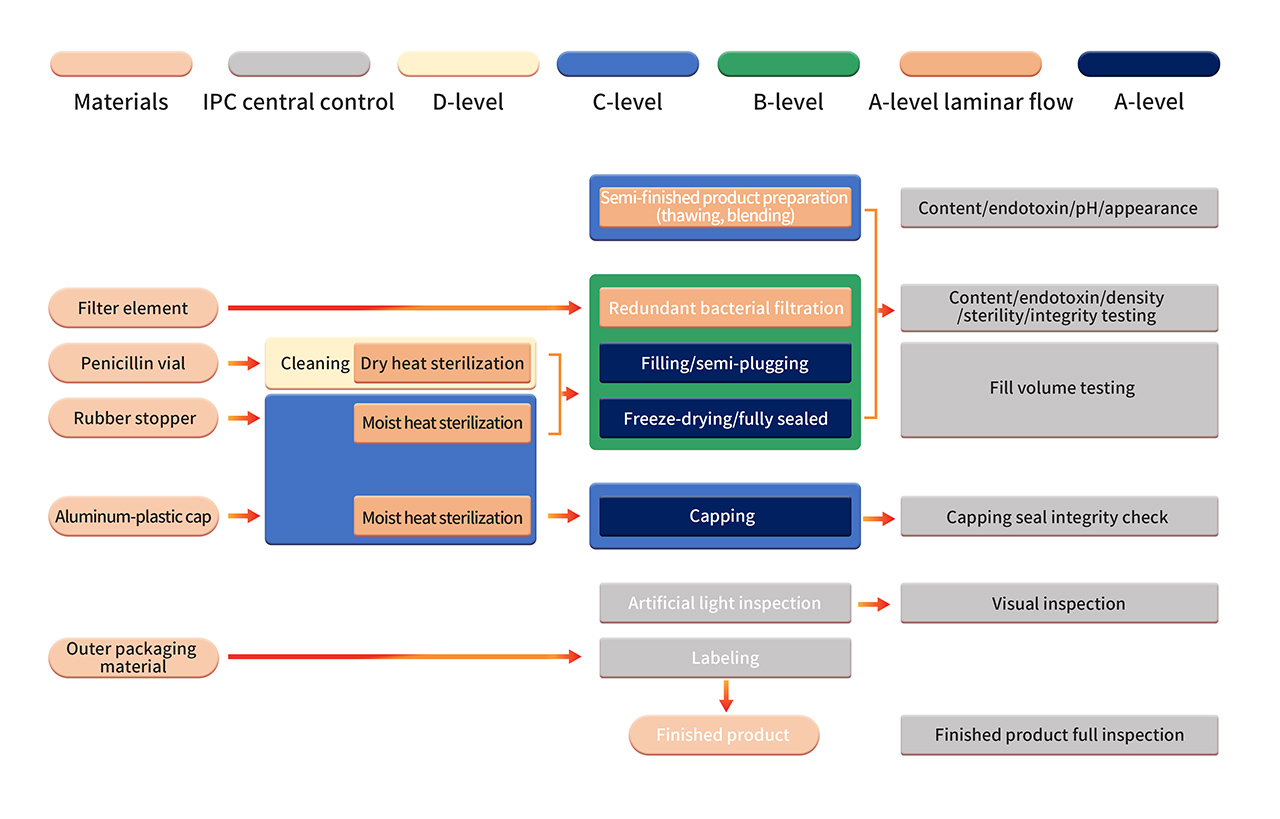
7. Strict aseptic process validation includes regular confirmation of aseptic process control, including the use of aseptic nutrient media and/or product substitutes in APS (media simulation containers). APS evaluates all aseptic operations conducted from sterilization and cleaning of process materials to container sealing. Various known aseptic operations and interventions under normal production and worst-case conditions are considered.
Reference regulation:
• EU GMP Annex 1.
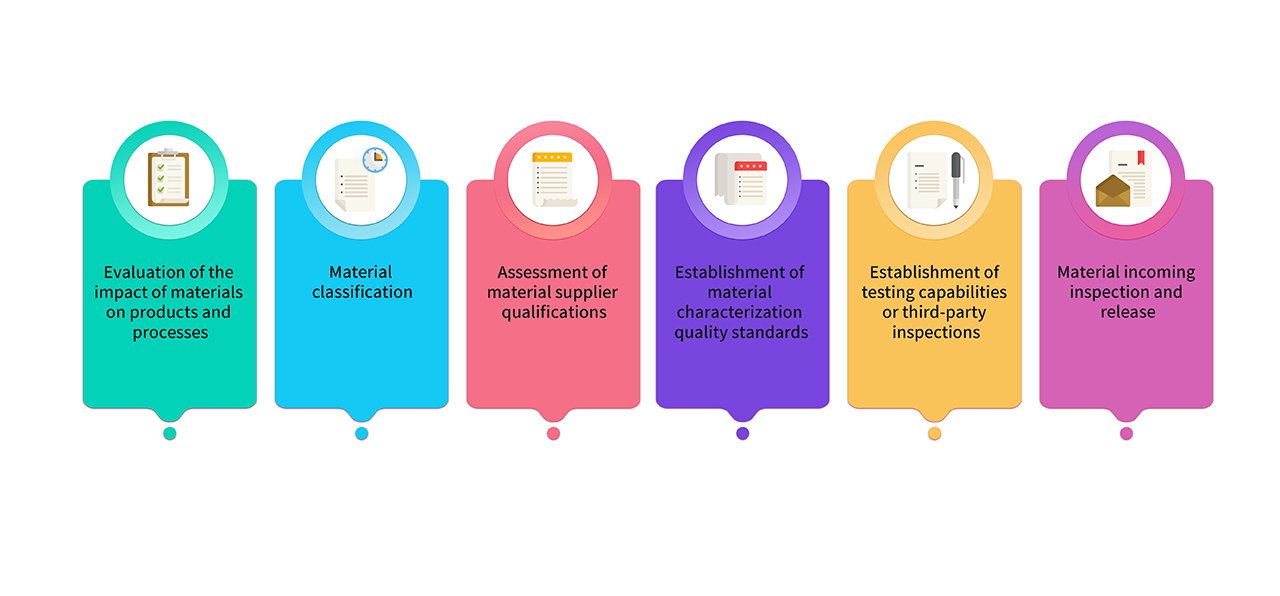
8. Raw materials and packaging materials used for production are selected from pharmaceutical-grade raw materials. For materials used in GMP product production, they undergo joint evaluation and classification by research, production, and quality teams. Key materials comply with the requirements of relevant regulations and pharmacopoeias, and their quality standards are established through critical characterization analysis. The material management process is as follows :
9. Our GMP-grade products are not only produced under a pharmaceutical-grade manufacturing plant but are also designed and controlled under relevant domestic and international CGT regulations. Taking the GMP-grade IL-15 as an example, our final product quality control release includes the followings:
• SDS-PAGE>95%
• Endotoxin level less than 10 EU/mg
• Residual Host Cell DNA content less than 0.02ng/μg
• Residual Host Cell Protein content less than 0.5ng/ug
• Biological activity >0.8 x 107 IU/mg (Reference the WHO Human IL-15 (NIBSC code: 90/530) as standard)
• Microbial testing
• Mycoplasma testing
• In vitro virus assay
• Batch-to-batch consistency
• Comprehensive stability data support (accelerated, freeze-thaw, long-term, shipping stability verification)
ACROBiosystems strictly controls potential exogenous factors throughout the entire process of development, production, quality testing, and quality system, ensuring the safety of our products. We are committed to developing high-quality reagents for clinical use in cell and gene therapy and combining drug production standards with stricter quality management and release testing standards to provide customers with bettersafer GMP grade products!
GMP Product Quality In-Depth Interpretation Special Series Trailer is Here!
• Controlling external contaminants in the production of critical materials for cell and gene therapies.
• Aseptic Protection Strategies for Critical Material Production for CGT
• Quality Control System for Critical Materials in CGT
• Global Supply Chain Security System for Critical Materials in CGT
• How to Better Meet Regulatory Requirements of the United States for Critical Materials in CGT
• ...







This web search service is supported by Google Inc.
















 A-Z
A-Z